
Vsebina
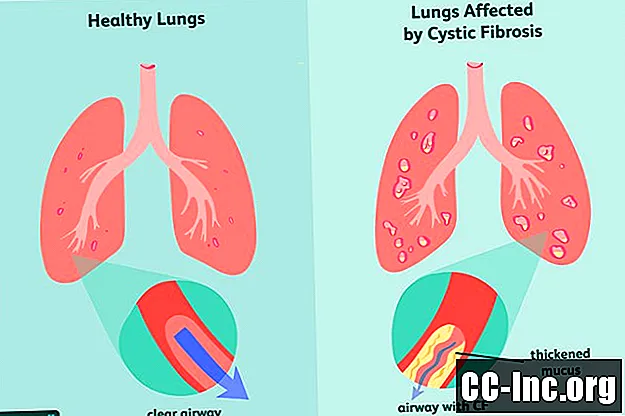
Cistična fibroza (CF) je dedna, življenjsko nevarna motnja, ki poškoduje pljuča in prebavni trakt. Povzroča ga okvarjen gen, ki sproži nastajanje zgoščene sluzi, ki zamaši dihalne poti in blokira izločanje prebavnih encimov.Simptomi so progresivni in pogosto hudi in lahko vključujejo težave z dihanjem, ponavljajoče se okužbe pljuč, slabo rast, moško neplodnost in kronično vnetje trebušne slinavke, jeter, ledvic in srca.
CF lahko diagnosticiramo s preiskavami krvi, genetskim pregledom in postopkom, znanim kot test znojnega klorida.
Čeprav CF ni zdravila, obstajajo načini zdravljenja, ki lahko izboljšajo dolžino in kakovost življenja.
Sem spadajo tehnike čiščenja dihalnih poti, inhalacijski antibiotiki, sredstva za redčenje sluzi, encimi trebušne slinavke, visokokalorična dieta in zdravila nove generacije, znana kot CFTR modulatorji. V hujših primerih bo morda potrebna presaditev pljuč.
Simptomi cistične fibroze
Kot genetska motnja je cistična fibroza nekaj, s čimer se rodiš. Ob rojstvu se lahko kaže ali ne, simptomi pa lahko trajajo mesece ali celo leta, preden se pojavijo znaki bolezni. Takrat so pljuča in prebavni trakt morda že imeli poškodbe, ki jih ni mogoče odpraviti.
Najpogostejši zgodnji znaki in simptomi CF vključujejo:
- Blokada otrokovega prvega blata (mekonij)
- Koža slanega okusa
- Kronični kašelj, sopenje ali obarvan sputum
- Ohlapno, mastno in običajno smrdljivo blato
- Okužba pljuč, pogosto ponavljajoča se
- Slaba rast in neuspeh
Če teh simptomov ni mogoče nadzorovati, ima lahko stres na pljučih (in nezmožnost pridobivanja teže) kumulativni učinek, ki prizadene več organov in poveča tveganje za zaplete bolezni.
Nekateri bolj značilni zapleti vključujejo:
- Zakasnjena puberteta
- Bronhiektazije (kronično zadebelitev pljučnih sten)
- Izguba teže
- Pankreatitis (vnetje trebušne slinavke)
- Moška neplodnost
- Pljučna hipertenzija (visok krvni tlak v pljučih)
- Žolčni kamni
- Sladkorna bolezen, povezana s cistično fibrozo
- Cor pulmonale (desnostransko srčno popuščanje)
- Ciroza (funkcionalno brazgotinjenje jeter)
Ker CF povzroča progresivne poškodbe celic in tkiv, bo vsaka škoda na pljučih in drugih organih v veliki meri nepopravljiva. Smrt je najpogosteje posledica dihalne odpovedi, sledi srčno popuščanje in odpoved jeter.
Simptomi cistične fibroze
Vzroki
Cistično fibrozo povzroča mutacija gena za transmembranski receptor za cistično fibrozo (CFTR), ki je odgovoren za proizvodnjo beljakovin CFTR. . Če je beljakovina deformirana ali je okvarjena, lahko povzroči dehidracijo na površini celice in povzroči zgostitev sluzi v okolici.
CF je avtosomno recesivna motnja, kar pomeni, da morate za bolezen podedovati mutacijo CFTR od matere in očeta. Če podedujete samo en okvarjen gen, ne boste imeli CF, temveč boste nosilec mutiranega gena.
Bolezen lahko podedujete, če ima vsak od staršev mutacijo CFTR ali sam CF. Če sta oba starša prevoznika, bi imeli:
- 25-odstotna možnost za CF
- 50-odstotna možnost, da postanete prevoznik
- 25-odstotna verjetnost, da nanj ne vplivate
Po drugi strani pa, če je eden od vaših staršev prevoznik, drugi pa ima CF, imate 50/50 možnosti, da imate CF ali ste prevoznik.
Cistična fibroza je ena najpogostejših genetskih bolezni, ki prizadene približno vsakega 2.500 dojenčkov, rojenih v ZDA.
Najpogostejša je med belci in Hispanicami, manj pogosto pa se pojavlja pri ljudeh afriškega ali azijskega porekla.
Dejavniki tveganja za cistično fibrozoDiagnoza
Za diagnosticiranje cistične fibroze se uporablja nekaj testov. Delujejo bodisi z neposrednim odkrivanjem mutacije CFTR bodisi posredno z merjenjem bioloških sprememb v skladu z boleznijo. Način diagnoze se lahko med nosečnostjo, rojstvom otroka ali kadar koli pozneje razlikuje.
Vodnik za razprave o zdravniku cistične fibroze
Pri naslednjem zdravniškem sestanku si oglejte naš vodnik za tiskanje, ki vam bo pomagal zastaviti prava vprašanja.

Od dveh običajnih testov, ki se običajno uporabljajo za diagnosticiranje CF:
- Preskušanje znojnega klorida, znan tudi preprosto kot test znoja, meri količino klorida na koži. Ker CF moti prenos soli v celice in iz njih, se bo sol kopičila v znoju.
- Genetsko testiranje CFTR se uporablja za odkrivanje najpogostejših mutacij mutacije CFTR. Čeprav obstaja več kot 2000 mutacij CFTR, za katere je znano, da povzročajo cistično fibrozo, 23, vključenih v standardni panel, predstavlja najverjetnejše osumljence.
Med nosečnostjo se lahko z genetskim testom CFTR testirajo tekočine, pridobljene z amniocentezo, ali celice, pridobljene z vzorčenjem horionskih resic (CVS).
Presejanje novorojenčkov se običajno uporablja tudi za diagnosticiranje CF in je danes pooblaščen v vseh 50 zveznih državah in okrožju Columbia. Kaj to pomeni, se bo razlikovalo glede na to, kje v ZDA živite. Če so rezultati presejalnega testiranja za novorojenčka pozitivni, se za potrditev diagnoze uporabi test znoja.
Kako se diagnosticira cistična fibrozaZdravljenje
Čeprav cistične fibroze ni mogoče pozdraviti, je napredek v zdravljenju podaljšal življenjsko dobo tistih, ki živijo s to boleznijo.
Cilj zdravljenja CF je štirikrat: preprečiti okužbe, ohraniti pljučno funkcijo, normalizirati prebavo in upočasniti napredovanje bolezni.
Med terapevtskimi orodji za zdravljenje CF:
- Tehnike čiščenja dihalnih poti (ACT) se izvajajo za odstranitev in izločanje nakopičene sluzi iz pljuč. Tehnike vključujejo močan kašelj, tolkala v prsih ali nihanje stene prsnega koša.
- Dieta z visoko vsebnostjo maščob in visoko kalorijami se uporablja za kompenzacijo malabsorpcije maščob, beljakovin in hranil v črevesju.
- Dodatek encimov trebušne slinavke se uporabljajo za krepitev prebavnih encimov, ki jih trebušna slinavka ne more proizvajati zaradi prekomernega kopičenja sluzi.
- Antibiotiki jemljejo vsak dan za preprečevanje bakterijskih okužb pljuč.
- Mukolitiki-se lahko uporabljajo zdravila za redčenje sluzi pred ACT.
- CFTR modulatorji so nov razred zdravil, ki lahko odpravijo določene napake v beljakovini CFTR in obnovijo njihovo regulacijsko funkcijo.
- Terapija s kisikom se lahko uporablja med akutnimi epizodami, ko je dihanje močno okvarjeno.
- Enteralna prehrana, znano tudi kot hranjenje po cevki, se lahko uporablja, če z normalno prehrano ne morete vzdrževati teže.
- Presaditev pljuč se šteje, ko vaša pljuča ne morejo več preživeti brez mehanskega prezračevanja
Spopadanje
Leta 1938, ko so cistično fibrozo prvič uvrstili med bolezni, so otroci redko živeli po prvem letu življenja. Do osemdesetih let bi lahko pričakovali, da bodo živeli od 20 do 25 let. Danes se je slika popolnoma spremenila pri ljudeh, ki živijo že v 40. in celo 50. letih, če se zdravljenje začne zgodaj in se ga drži.
To ne pomeni, da je CF nič manj resen kot kdaj koli prej. Je življenjski dogodek, ki zahteva skrbnost in doslednost, da se ne le spopade z boleznijo, temveč živi tudi najvišji možni življenjski standard.
V ta namen morate v svojem življenju normalizirati CF z vzpostavitvijo rutin in praks, da se izognete vzponom in padcem, ki lahko povzročijo stres in povečajo invalidnost. Med premisleki bi morali:
- Upravljajte s svojo prehrano. Ljudje s CF pogosto potrebujejo dvakrat dnevno kalorije kot drugi.
- Redno telovadi. V idealnem primeru bi morali fitnes rutine vključevati najmanj 20 do 30 minut aerobnih aktivnosti trikrat na teden. Poiščite nekaj prijetnega, kar lahko počnete celo življenje.
- Hranite dobro hidrirano. S tem pljuča in črevesje delujejo pravilno. Odvisno od starosti morate na dan spiti najmanj šest do osem visokih kozarcev vode.
- Pravilno izvedite zračnost. Ko se vaše zdravstvene potrebe spreminjajo, se lahko spremenijo tudi vrste orodij, ki jih potrebujete. Če ne dosežete želenih rezultatov, se pogovorite s svojim pulmologom ali fizioterapevtom.
- Poiščite podporo. Poleg prijateljev in družine se lahko obrnete na najbližje poglavje Fundacije za cistično fibrozo (CFF), da se vključite v omrežje za podporo na vašem območju.
- Poiščite finančno pomoč. CFF ponuja storitve, ki družinam pomagajo, da se lažje spopadejo z visokimi stroški zdravljenja CF.
Beseda iz zelo dobrega
Medtem ko so presejanja novorojenčkov dramatično povečala stopnjo diagnoz CF pri dojenčkih, več kot 25 odstotkov diagnoz postavijo le v otroštvu, najstniških letih in zgodnjih odraslih letih.
To je problematično, saj lahko zgodnja diagnoza in zdravljenje preprečita številne hujše zaplete CF, preden je mogoče narediti resnejšo škodo. Čeprav zdravljenje ne more ustaviti ali obrniti bolezni, lahko pa zagotovi še veliko let brez bolezni.
V ta namen je pomembno poznati zgodnje simptome CF in se pogovoriti s svojim zdravnikom, če sumite, da ima vaš otrok bolezen. To še posebej velja za države, ki pregledujejo samo krvne preiskave IRT, kar bi lahko imelo za posledico pri približno 5 odstotkih otrok bodisi zapoznelo diagnozo bodisi lažno negativni rezultat, kažejo raziskave na Medicinski fakulteti za javno zdravje Univerze v Wisconsinu. .
Katere simptome lahko pričakujete pri cistični fibrozi?