
Vsebina
- Vzroki
- Simptomi
- Izpiti in testi
- Zdravljenje
- Skupine za podporo
- Outlook (Prognoza)
- Možne zaplete
- Kdaj se obrnite na zdravnika
- Preprečevanje
- Alternativna imena
- Navodila za bolnika
- Slike
- Reference
- Datum pregleda 19.2.2018
Cistična fibroza je bolezen, ki povzroča nastanek debele, lepljive sluzi v pljučih, prebavnem traktu in drugih delih telesa. Je ena najpogostejših kroničnih pljučnih bolezni pri otrocih in mladih odraslih. To je življenjsko nevarna motnja.
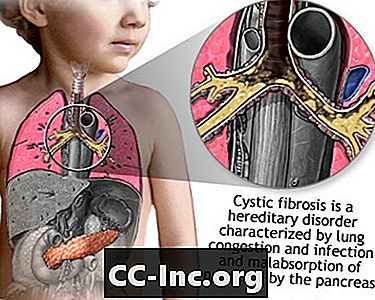
Vzroki
Cistična fibroza (CF) je bolezen, ki se prenaša skozi družine. Povzroča ga okvarjen gen, zaradi katerega telo proizvaja nenormalno debelo in lepljivo tekočino, imenovano sluz. Ta sluz se kopiči v dihalnih poteh pljuč in trebušne slinavke.
Nastajanje sluzi povzroči življenjsko nevarne okužbe pljuč in resne težave s prebavo. Bolezen lahko vpliva tudi na znojne žleze in moški reproduktivni sistem.
Mnogi ljudje nosijo gen CF, vendar nimajo simptomov. Razlog za to je, da mora oseba s CF podedovati 2 defektne gene, od katerih je vsak eden od staršev. Nekateri beli Američani imajo gen CF. Bolj je med tistimi severnega ali srednjeevropskega porekla.
Večina otrok s CF je diagnosticirana po starosti 2. Za majhno število se bolezen ne ugotovi do 18. leta starosti. Ti otroci imajo pogosto blažjo obliko bolezni.
Simptomi
Simptomi pri novorojenčkih lahko vključujejo:
- Zapoznela rast
- Nenavadno povečanje telesne mase v otroštvu
- V prvih 24 do 48 urah življenja ni gibanja črevesja
- Koža s slanim okusom
Simptomi, povezani s funkcijo črevesja, lahko vključujejo:
- Bolečine trebuha zaradi hude zaprtosti
- Povečan plin, napihnjenost ali trebuh, ki se zdi otekel (razširjen)
- Slabost in izguba apetita
- Stoli, ki so bledi ali glinaste barve, imajo neprijeten vonj, imajo sluz ali plavajo
- Izguba teže
Simptomi, povezani s pljuči in sinusi, lahko vključujejo:
- Kašelj ali povečana sluz v sinusih ali pljučih
- Utrujenost
- Zamašen nos, ki ga povzročajo nosni polipi
- Ponavljajoče se epizode pljučnice (simptomi pljučnice pri nekom s cistično fibrozo vključujejo zvišano telesno temperaturo, povečan kašelj in zasoplost, povečano sluz in izgubo apetita)
- Sinusna bolečina ali pritisk zaradi okužbe ali polipov
Simptomi, ki se lahko pojavijo pozneje v življenju:
- Neplodnost (pri moških)
- Ponavljajoče vnetje trebušne slinavke (pankreatitis)
- Respiratorni simptomi
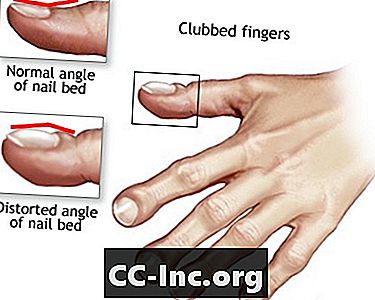
- Prsti na klupi
Izpiti in testi
Naredite krvni test, ki pomaga odkriti CF. Test išče spremembe v genu CF. Drugi testi za diagnozo CF vključujejo: t
- Test imunoreaktivnega tripsinogena (IRT) je standardni presejalni test za novorojenčka za CF. Visoka raven IRT predlaga možen CF in zahteva nadaljnje testiranje.
- Preskus znojnega klorida je standardni diagnostični test za CF. Visoka vsebnost soli v znoju osebe je znak bolezni.
Drugi testi, ki identificirajo težave, ki so lahko povezani s CF, vključujejo:
- Rentgenska slika prsnega koša ali CT
- Preskus fekalne maščobe
- Preskusi delovanja pljuč
- Merjenje funkcije pankreasa
- Test stimulacije sekretina
- Tripsin in kimotripsin v blatu
- Serija zgornje GI in črevesja
Zdravljenje
Zgodnja diagnoza CF in načrt zdravljenja lahko izboljšata preživetje in kakovost življenja. Spremljanje in spremljanje sta zelo pomembna. Kadar je mogoče, je treba skrbeti za specialno kliniko za cistično fibrozo. Ko otroci dosežejo odraslo dobo, se morajo preusmeriti v center specialnosti za cistično fibrozo za odrasle.
Zdravljenje pljučnih težav vključuje:
- Antibiotiki za preprečevanje in zdravljenje okužb pljuč in sinusov. Lahko jih jemljete peroralno ali v žilah ali z dihanjem. Osebe s CF lahko jemljejo antibiotike samo kadar je to potrebno ali ves čas. Odmerki so pogosto višji od običajnih.
- Vdihavanje zdravil za pomoč pri odpiranju dihalnih poti.
- Drugi zdravili, ki se dajo v dihalno zdravljenje tanki sluzi in olajšajo kašelj, so encim DNAse. ter visoko koncentrirane raztopine soli (hipertonična slanica).
- Cepivo proti gripi in pnevmokokno polisaharidno cepivo (PPV) letno (vprašajte svojega zdravnika).
- Presaditev pljuč je v nekaterih primerih možnost.
- Zdravljenje s kisikom je lahko potrebno, ker se bolezen pljuč poslabša.
Tudi težave s pljuči se zdravijo z zdravljenjem, da se sluz razreže. To olajša kašljanje sluzi iz pljuč.
Te metode vključujejo:
- Aktivnost ali vadba, ki povzroči globoko dihanje
- Naprave, ki se uporabljajo čez dan, da pomagajo očistiti dihalne poti preveč sluzi
- Ročno perkusijo prsnega koša (ali fizioterapija v prsnem košu), v kateri družinski član ali terapevt rahlo ploska osebo na prsih, hrbtu in območju pod rokami
Zdravljenje težav s črevesjem in prehranjevanjem lahko vključuje:
- Posebna dieta z visoko vsebnostjo beljakovin in kalorij za starejše otroke in odrasle
- Encimi trebušne slinavke pomagajo absorbirati maščobe in beljakovine, ki jih jemljete z vsakim obrokom
- Vitaminski dodatki, zlasti vitamini A, D, E in K
- Če imate zelo trdo blato, vam lahko vaš zdravnik svetuje druga zdravljenja
Zdravilo Ivacaftor in Lumacaftor so zdravila, ki zdravijo določene vrste CF. Izboljšajo funkcijo enega od defektnih genov, ki povzročajo CF. Posledično je v pljučih manj gosto sluz. Tudi drugi simptomi CF so izboljšani.
Nega in spremljanje doma morata vključevati:
- Izogibanje dimu, prahu, umazaniji, param, gospodinjskim kemikalijam, dimnemu ognju in plesni ali plesni.
- Dajanje veliko tekočine, zlasti dojenčkom in otrokom v vročem vremenu, ko je driska ali ohlapno blato, ali med dodatno telesno dejavnostjo.
- Vadba 2 ali 3-krat na teden. Plavanje, tek in kolesarjenje so dobre možnosti.
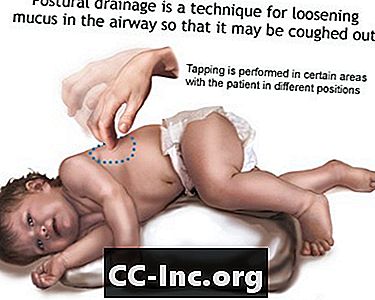
- Odstranjevanje sluzi ali izločkov iz dihalnih poti. To je treba opraviti 1 do 4-krat na dan. Pacienti, družine in negovalci se morajo naučiti izvajati tolkanje v prsih in posturalno drenažo, da bodo dihalne poti ostale jasne.
Skupine za podporo
Stres bolezni lahko ublažite tako, da se pridružite skupini za podporo cistične fibroze. Delitev z drugimi, ki imajo skupne izkušnje in težave, lahko vaši družini pomaga, da se ne počuti sam.
Outlook (Prognoza)
Večina otrok s CF ostane v dobrem zdravju, dokler ne doseže odrasle dobe. Sposobni so sodelovati pri večini dejavnosti in se udeležiti šole. Mnogi mladi odrasli s CF končajo šolo ali najdejo zaposlitev.
Bolezen pljuč se na koncu poslabša do točke, ko je oseba invalidna. Danes je povprečna življenjska doba ljudi s CF, ki živijo v odrasli dobi, približno 37 let.
Smrt je najpogosteje posledica zapletov pljuč.
Možne zaplete
Najpogostejši zaplet je kronična okužba dihal.
Drugi zapleti so:
- Težave s črevesjem, kot so žolčni kamni, črevesna blokada in rektalni prolaps
- Kašelj krvi
- Kronična dihalna odpoved
- Diabetes
- Neplodnost
- Bolezni jeter ali odpovedi jeter, pankreatitis, žolčnica
- Podhranjenost
- Nosni polipi in sinusitis
- Osteoporoza in artritis
- Pljučnica, ki se vedno znova vrača
- Pneumotoraks
- Desna stranska srčna odpoved (cor pulmonale)
Kdaj se obrnite na zdravnika
Pokličite svojega ponudnika, če ima otrok ali otrok simptome CF in izkušnje:
- Povišana telesna temperatura, povečan kašelj, spremembe v izpljunku ali krvi v izpljunku, izguba apetita ali drugi znaki pljučnice
- Povečana izguba teže
- Pogostejša črevesna gibanja ali blato, ki je neprijeten ali ima več sluzi
- Otekel trebuh ali povečano napihnjenost
Pokličite svojega ponudnika, če se pri osebah s CF razvijejo novi simptomi ali če se simptomi poslabšajo, zlasti hude težave pri dihanju ali kašljanje krvi.
Preprečevanje
CF ni mogoče preprečiti. Preskušanje tistih z družinsko anamnezo bolezni lahko pri mnogih nosilcih odkrije gen CF.
Alternativna imena
CF
Navodila za bolnika
- Enteralna prehrana - otrokove težave
- Cevka za gastrostomsko hranjenje - bolus
- Kako dihati, ko vam je težko dihati
- Jejunostomska cev za hranjenje
- Posturalna drenaža
Slike
Clubbing
Posturalna drenaža
Prsti na klupi
Cistična fibroza
Reference
Accurso FJ. Cistična fibroza. V: Goldman L, Schafer AI, eds. Medicina Goldman-Cecil. 25. izd. Philadelphia, PA: Elsevier Saunders; 2016: poglavje 89.
Egan ME, Green DM, Voynow JA. Cistična fibroza. V: Kliegman RM, Stanton BF, St. Geme JW, Schor NF, eds. Nelsonov učbenik za pediatrijo. 20. ed. Philadelphia, PA: Elsevier; 2016: pogl. 403.
Farrell PM, White TB, Ren CL, et al. Diagnoza cistične fibroze: smernice za soglasje fundacije Cystic Fibrosis. J Pediatr. 2017; 181S: S4-S15.e1. PMID: 28129811 www.ncbi.nlm.nih.gov/pubmed/28129811.
Rowe SM, Hoover W, Solomon GM, Sorscher EJ. Cistična fibroza. V: Broaddus VC, Mason RJ, Ernst JD, et al, eds. Murray in Nadelov učbenik za respiratorno medicino. 6. izd. Philadelphia, PA: Elsevier Saunders; 2016: poglavje 47.
Taylor-Cousar JL, Munck A, McKone EF, et al. Tezacaftor-ivacaftor pri bolnikih s cistično fibrozo homozigotnih za phe508del. N Engl J Med. 2017; 377 (21): 2013–2023. PMID: 29099344 www.ncbi.nlm.nih.gov/pubmed/29099344.
Datum pregleda 19.2.2018
Posodobljeno: Neil K. Kaneshiro, MD, MHA, klinični profesor pediatrije, Medicinska fakulteta Univerze v Washingtonu, Seattle, WA. Pregledali so ga tudi David Zieve, MD, MHA, medicinski direktor, Brenda Conaway, urednica in A.D.A.M. Uredniška ekipa.